Retinitis Pigmentosa

Retinitis pigmentosa (RP) is a group of diseases which tend to run in families that cause progressive degeneration of the retina in both eyes. It progresses from night blindness to loss of the peripheral visual field. The signs of the disease usually present in youth or young adulthood, but can occur at any age. Over many years, RP then progresses to tunnel vision and finally blindness.
In approximately half of all cases (50 to 60%) there are other family members with RP. It affects approximately 1 in 3,000 to 4,000 people.
Difficulties with night vision and peripheral vision are the first things that are noticed. Later, reading vision and colour vision are affected. The age at which symptoms start is variable and may vary with the different genetic types. Progression of RP is different in each case.
RP is a type of hereditary retinal dystrophy, a group of inherited disorders in which abnormalities of the photoreceptors (rods and cones) or the retinal pigment epithelium (RPE) of the retina lead to progressive visual loss.
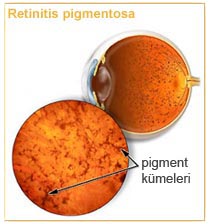
Someone with retinitis pigmentosa will notice gradual changes in vision, including difficulty seeing at night, loss of peripheral vision (donut shaped visual field loss) and sensation of twinkling or flashing light. Night blindness refers to an inability to adjust vision to the darkness or a very slow adjustment. During later stages, the next symptom to appear is a gradual loss of peripheral or side vision so that the person can only see through a small area of the eye and only straight ahead, this is also referred to as tunnel vision.
Retinitis pigmentosa usually affects both eyes. In some forms of the condition, vision continues to get worse. In other types of retinitis pigmentosa, only a small area is affected and vision might not change at all for several years.
rme alanında ise genellikle çevresel daralma görülür ve tünelden bakıyormuş hissi oluşur. Bu nedenle hastalar etraftaki eşyaları farkedemezler ve sık kazaya sebep olurlar..
The diagnosis of retinitis pigmentosa relies upon documentation of progressive loss in photoreceptor function by electroretinography (ERG) and visual field testing. The mode of inheritance of RP is determined by family history.
RP can be inherited in an autosomal dominant, autosomal recessive, or X-linked manner.
Genetic counseling depends on an accurate diagnosis, determination of the mode of inheritance in each family, and results of molecular genetic testing.
RP combined with progressive deafness is called Usher syndrome.
Unfortunately, there is no effective treatment to stop or cure retinitis pigmentosa. Research is being conducted, but there is currently not even an experimental medication or surgical treatment.
The progression of the disease can be reduced by the daily intake of vitamin A
Last Updated: October 23, 2024